(Note: This post is very much a work in progress. I’m going through and rewriting a script with my new python skills I have been learning)
In order to use Molecular Dynamics (MD) simulations to determine the energy of water within a protein, water actually needs to be inside the protein. However, starting coordinates of proteins typically do not contain water.
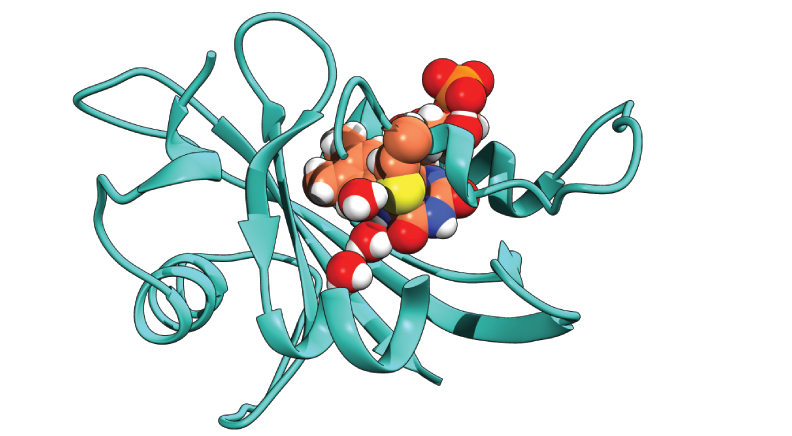
I wrote some code to take simulation data in text form containing position and momentum in 3 dimensions for all atoms (the .gro file format from the GROMACS MD suite to be precise), ask the user to pick 3 residues and an atom from all 3, then insert water into every frame at the middle point between 3 residues. The pictures in this post were made after utilizing the code to add water to the interior of the protein model. I made the images from simulation frames visualized in UCSF Chimera, then processed with photoshop and illustrator.

I wrote this script as I was learning python. Since then I’ve learned a lot more. When I reread it before posting here I found some things I would change if I were to write it again today. Here are some snippets I rewrote. The full script is at the bottom of the post.
I took this large and semi-readable block and rewrote it by defining a couple functions, and renaming some variables to be more understandable

The new functions:

And this made the code much cleaner, and a bit more concise.

It’s still not perfect, as the loops on lines 80 and 75 have a hardcoded number to iterate up to. I haven’t done the math to figure out a function output the right number yet.
Here’s the original code:
# script to create acceptable .gro file for processing into tpic inp trajectory from .gro format trajectory
import sys
import math
import os
# import file and read each line into list
print "importing .gro file"
trj_out = open(sys.argv[2], 'w')
trj_raw = open(sys.argv[1], 'r' )
list_lines = trj_raw.read().splitlines()
trj_raw.close()
# get input from user to pick which amino acids and atoms in the amino acids to put the water between
aa_1_no = raw_input( "which amino acid to take first coordinates from? Please use the form 100ALA, and input amino acids in numerical order, ie amino acid 13 should be imputted before amino acid 14) ")
aa_1_atom_no = raw_input( "which atom in that amino acid to take coordinate of? (please use caps) ")
aa_2_no = raw_input( "which amino acid to take second coordinates from? (please use the form 100ALA) ")
aa_2_atom_no = raw_input( "which atom in that amino acid to take coordinate of? (please use caps) ")
aa_3_no = raw_input( "which residue to take third coordinates from? (use the form 103ALA) ")
aa_3_atom_no = raw_input( "which atom in that residue to take coordinates of? (use caps) ")
# some test atoms and amino acids hard coded
#aa_1_no = '103CYS'dd
#aa_1_atom_no = 'S'
#aa_2_no = '135CYS'
#aa_2_atom_no = 'S'
# some housekeeping to keep the correct format of the input and output files
# must make sure the correct number of particles, and frames is written
old_particle_no = list_lines[1]
new_particle_no = int(old_particle_no) + 1
new_particle_no = repr(new_particle_no)
print new_particle_no
print 'old particle number = {} , new particle number = {} '.format(old_particle_no, new_particle_no)
no_of_original_lines = len(list_lines)
no_of_frames = list_lines.count(old_particle_no)
lines_per_frame = no_of_original_lines / no_of_frames
print "number of frames = {} ".format(no_of_frames)
old_last_residue_no = list_lines[ ( no_of_original_lines - 2 ) ].translate(None, 'CLNASOL').split()[0]
new_last_residue_no = int(old_last_residue_no) + 1
print 'original last residue number = {} , new last residue number = {} '.format(old_last_residue_no, new_last_residue_no)
# make generator from the list composed of the lines in the input file
gen = (n for n,y in enumerate(list_lines))
# iterate over generator to make individual strings of each list member (ie each line in the list is now a string)
for n in gen:
line_contents_a = list_lines[n-1:n]
line_str_a = ', '.join(map(str, line_contents_a))
#print 'line number = {} line contents = {}'.format(n, line_contents)
#if line_n is '12824' :
# print 'this is line_str {}'.format(line_str)
# change metadata at top of new file to reflect new particle numbers
if line_str_a == str(old_particle_no) :
print "changing particle number"
list_lines[n-1] = new_particle_no
# find the lines containing the first of the atoms between which the new water will be inserted
if line_str_a.startswith( aa_1_no , 2 ) and line_str_a.startswith( aa_1_atom_no , 13 ):
# print the frame number being modified. mostly just for entertainment but the frame number itself is needed later
for x in range(1, (no_of_frames + 1 )) :
#print 'reading the first {} lines '.format(x * lines_per_frame)
if (x * lines_per_frame) > n :
current_frame_no = x
print 'current frame is {} '.format(current_frame_no)
break
atom_a_line = line_str_a
print "atom a line = {} ".format(atom_a_line)
# now time to find the next atom from the next amino acid. I think the amino acids need be inputted in order that they appear for this block to work
for m in range(1, 1000000):
line_contents_b = list_lines[ m+n-1 : m+n ]
line_str_b = ', '.join(map(str, line_contents_b))
if line_str_b.startswith( aa_2_no , 2 ) and line_str_b.startswith( aa_2_atom_no , 13 ):
atom_b_line = ', '.join(map(str, list_lines[n+m-1 : m+n]))
print 'atom b line = {} '.format(atom_b_line)
#now its time to rip the strings apart to get the tasty coordinate data.
segments_a = atom_a_line.split()
segments_b = atom_b_line.split()
# since the third amino acid/atom to place the water between as added later I put the code to match the third atom after
# the line.split() call. Possible I defined segements_a and segments_b twice for no good reason
for p in range(1, 1000000):
line_contents_c = list_lines[ m + n + p - 1 : m + n + p ]
line_str_c = ', '.join(map(str, line_contents_c))
if line_str_c.startswith( aa_3_no , 2 ) and line_str_c.startswith( aa_3_atom_no , 13 ):
atom_c_line = ', '.join(map(str, list_lines[n+m-1 : m+n]))
print 'atom b line = {} '.format(atom_c_line)
segments_a = atom_a_line.split()
segments_b = atom_b_line.split()
segments_c = atom_c_line.split()
#define current and new xyz coords after extracting the correct slice from the line.split and turning it into float
a_x = float(segments_a[3])
a_y = float(segments_a[4])
a_z = float(segments_a[5])
b_x = float(segments_b[3])
b_y = float(segments_b[4])
b_z = float(segments_b[5])
c_x = float(segments_c[3])
c_y = float(segments_c[4])
c_z = float(segments_c[5])
# math to define point in between the 3 atoms
newx = round((a_x+b_x)/2, 3)
newy = round((a_y+b_y)/2, 3)
newz = round((a_z+b_z)/2, 3)
# new string to be jammed into end of frame.
new_coord = '{}SOL {} {} {} {}'.format(new_last_residue_no, new_particle_no, newx, newy, newz )
print 'new line from average coordinates = {} '.format(new_coord)
# determine where to insert new line based on frame number and lines per frame determined at beginning of script, insert line and break back to the next frame.
insert_new_coord_at = ( current_frame_no * lines_per_frame )-1
list_lines.insert(insert_new_coord_at, new_coord)
break
# add line to close files after reading? no, closed raw input file at beginning of script.
print ' writing output file, " {} " '.format(sys.argv[2])
trj_out.write('\n'.join(list_lines))
print "complete."
# if line.startswith(' 449ASN'):
#ASN_line = line
#VAL_line = line - 533
#ASN_line.splitA
And the new version:
<span id="mce_SELREST_start" style="overflow:hidden;line-height:0;"></span>
# script to create acceptable .gro file for processing into tpic inp trajectory from .gro format trajectory# script to create acceptable .gro file for processing into tpic inp trajectory from .gro format trajectory
import sysimport mathimport os
def list_slice_2_str(lis, number): line_contents_a = list_lines[number - 1: number] line_str = ', '.join(map(str, line_contents_a)) return line_str
def return_frame_no(number): for x in range(1, (no_of_frames + 1)): if (x * lines_per_frame) > number: current_frame_no = x return current_frame_no
# import file and read each line into listprint "importing .gro file"# trj_out = open(sys.argv[2], 'w')# trj_raw = open(sys.argv[1], 'r' )trj_out = open('test_out.gro', 'w')trj_raw = open('tpic_test.gro', 'r')
list_lines = trj_raw.read().splitlines()trj_raw.close()aa_1_no = '103CYS'aa_1_atom_no = 'S'aa_2_no = '104ASP'aa_2_atom_no = 'C'aa_3_no = '105ILE'aa_3_atom_no = 'C'# get input from user to pick which amino acids and atoms in the amino acids to put the water between# aa_1_no = raw_input( "which amino acid to take first coordinates from? Please use the form 100ALA, and input amino acids in numerical order, ie amino acid 13 should be imputted before amino acid 14) ")# aa_1_atom_no = raw_input( "which atom in that amino acid to take coordinate of? (please use caps) ")
# aa_2_no = raw_input( "which amino acid to take second coordinates from? (please use the form 100ALA) ")# aa_2_atom_no = raw_input( "which atom in that amino acid to take coordinate of? (please use caps) ")
# aa_3_no = raw_input( "which residue to take third coordinates from? (use the form 103ALA) ")# aa_3_atom_no = raw_input( "which atom in that residue to take coordinates of? (use caps) ")
# some housekeeping to keep the correct format of the input and output files # must make sure the correct number of particles, and frames is writtenold_particle_no = list_lines[1]new_particle_no = int(old_particle_no) + 1new_particle_no = repr(new_particle_no)print new_particle_noprint 'old particle number = {} , new particle number = {} '.format(old_particle_no, new_particle_no)no_of_original_lines = len(list_lines)no_of_frames = list_lines.count(old_particle_no)lines_per_frame = no_of_original_lines / no_of_framesprint "number of frames = {} ".format(no_of_frames)old_last_residue_no = list_lines[(no_of_original_lines - 2)].translate(None, 'CLNASOL').split()[0]new_last_residue_no = int(old_last_residue_no) + 1print 'original last residue number = {} , new last residue number = {} '.format(old_last_residue_no, new_last_residue_no)
# make generator from the list composed of the lines in the input file and indicesgen = (n for n, y in enumerate(list_lines))# iterate over generator to make individual strings of each list member (ie each line in the list is now a string)for iter_1_no in gen: line_str_a = list_slice_2_str(list_lines, iter_1_no) if line_str_a == str(old_particle_no): print "changing particle number" list_lines[iter_1_no - 1] = new_particle_no print 'line_str_a = {} '.format(line_str_a) if line_str_a.startswith(aa_1_no, 2) and line_str_a.startswith(aa_1_atom_no, 13): current_frame_no = return_frame_no(iter_1_no) print 'current frame is {}'.format(str(current_frame_no)) atom_a_line = line_str_a segments_a = atom_a_line.split() print "atom a line = {} ".format(atom_a_line) for iter_2_num in range(1, 1000000): line_str_b = list_slice_2_str(list_lines, iter_1_no + iter_2_num) if line_str_b.startswith(aa_2_no, 2) and line_str_b.startswith(aa_2_atom_no, 13): atom_b_line = line_str_b segments_b = atom_b_line.split() for iter_3_no in range(1, 1000000): line_str_c = list_slice_2_str(list_lines, iter_2_num + iter_1_no + iter_3_no) if line_str_c.startswith(aa_3_no, 2) and line_str_c.startswith(aa_3_atom_no, 13): atom_c_line = line_str_c print 'atom c line = {} '.format(atom_c_line) segments_c = atom_c_line.split() # define current and new xyz coords after extracting the correct slice from the line.split and turning it into float a_x = float(segments_a[3]) a_y = float(segments_a[4]) a_z = float(segments_a[5]) b_x = float(segments_b[3]) b_y = float(segments_b[4]) b_z = float(segments_b[5]) c_x = float(segments_c[3]) c_y = float(segments_c[4]) c_z = float(segments_c[5]) # math to define point in between the 3 atoms newx = round((a_x + b_x) / 2, 3) newy = round((a_y + b_y) / 2, 3) newz = round((a_z + b_z) / 2, 3) # new string to be jammed into end of frame. new_coord = '{}SOL {} {} {} {}'.format(new_last_residue_no, new_particle_no, newx, newy, newz) print 'new line from average coordinates = {} '.format(new_coord) # determine where to insert new line based on frame number and lines per frame determined at beginning of script, insert line and break back to the next frame. insert_new_coord_at = (current_frame_no * lines_per_frame) - 1 list_lines.insert(insert_new_coord_at, new_coord) break
# add line to close files after reading? no, closed raw input file at beginning of script.#print ' writing output file, " {} " '.format(sys.argv[2])trj_out.write('\n'.join(list_lines))print "complete."# if line.startswith(' 449ASN'):# ASN_line = line# VAL_line = line - 533# ASN_line.splitA
End! Thanks for reading
[…] after I produced MD simulations with a water within a protein, I needed both a quantification of whether the water […]